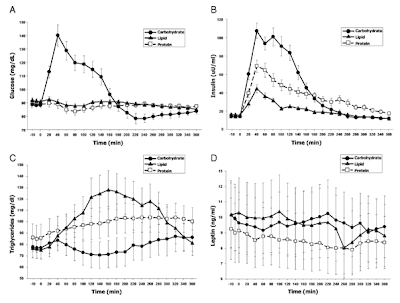
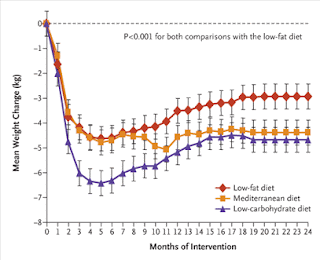
Fat storage in pancreas and in insulin-sensitive tissues in pathogenesis of type 2 diabetes
Fat storage in pancreas and in insulin-sensitive tissues in pathogenesis of type 2 diabetes Obesity is associated with increased storage of lipids in nonadipose tissues like skeletal muscle, liver, and pancreatic b cells. These lipids constitute a continuous source of long-chain fatty acyl CoA (LC-CoA) and derived metabolites like diacylglycerol and ceramide, acting as signalling molecules on protein kinases activities (in particular, the family of PKCs), ion channel, gene expression, and protein acylation. In skeletal muscle, the increase in LC-CoA and diacylglycerol translocates and activates specific protein kinase C (PKC) isoforms, which will phosphorylate IRS-1 on serine, preventing its phosphorylation on tyrosine and association with PI3 kinase. This interrupts the insulin signalling pathway leading to the stimulation of glucose transport. In pancreatic b cells, short-term excess of fatty acids or LC-CoA activates PKC and also directly stimulates insulin exocytosis. Longterm ex...