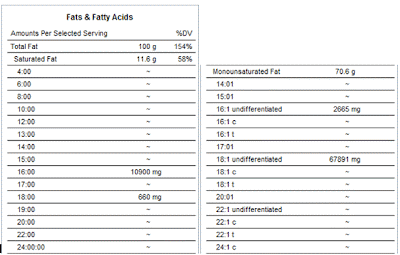
So let me start by saying that I've gleaned a lot of useful information over at Dr. Davis' Heart Scan Blog . But one thing I've noticed is that he rarely responds to comments left at his site. Now, obviously, he's under no obligation to do so, but when the comments are corrective in nature, not doing so leaves the impression that he didn't even bother to read the comments and/or take them under advisement. The first time I noticed this was in this post . Carbohydrates in the diet trigger formation of small LDL particles. Because carbohydrates, such as products made from wheat, increase triglycerides and triglyceride-containing lipoproteins ( chylomicrons, chylomicron remnants , VLDL, and IDL), LDL particles (NOT LDL cholesterol) become triglyceride-enriched. Triglyceride-enriched LDL particles are "remodeled" by the enzyme, hepatic lipase, into triglyceride-depleted, small LDL particles. I'll leave the rest of that alone and deal with just the ...