Hyperinsulinemia, Hyperglycemia and Insulin Resistance
Over in the comments on the Taubes Toothpick post, starting around here, Nostril Damus (funny!) challenged my contention that the only carb implicated in IR is fructose, a carb that doesn't elicit an insulin response. ND posited that other carbs cause hyperinsulinemia cause self-downregulation of insulin receptors leading to IR. Then Melchior posted about a study where hyperinsulinemia and hyperglycemia/insulinemia induced insulin resistance which provided more food for thought.
I thought on this a bit and think it is worthwhile to repeat in a post of its own and add a few more comments.
So a direct quotation of ND's premise: "Over time, this self-induced loss of target cell receptors for insulin reduces the target cell’s sensitivity to the elevated hormone concentration."
Unfortunately only the abstract is available free online, but it does contain some interesting information. Here's the study:
Two study protocols to examine the effects of chronic (72-96 h) physiologic euglycaemic hyperinsulinaemia (+ 72 pmol/l) and chronic hyperglycaemic (+ 1.4 mmol/l) hyperinsulinaemia (+ 78 pmol/l) on insulin sensitivity and insulin secretion were performed in 15 healthy young subjects.
It is important to recognize right off the bat here that we're talking healthy young subjects here with normal insulin sensitivity and through 3-4 day long infusions an artificial chronic state of hyperinsulinemia alone and hyperglycemia/insulinemia together was given. My point here being that we're not talking inducing an insulin spike, even a huge one, we're talking manipulating the insulin levels artificially. I also note we are talking about constantly elevated insulin and/or blood glucose levels. This situation may somewhat mimic what is present in T2 diabetics once metabolic havoc has been wrought, but it is simply not analogous in any manner to physiological insulin responses even to high doses of highly insulinogenic foods.
Subjects received a three-step euglycaemic insulin (insulin infusion rates = 1.5, 3, and 6 nmol.kg-1.min-1) clamp and a hyperglycaemia (6.9 mmol/l) clamp before and after chronic insulin or glucose infusion.
Following 4 days of sustained euglycaemic hyperinsulinaemia whole body glucose disposal decreased by 20-40%.
So IR here is assessed by whole body glucose disposal. The problem with this measure, and what scientists are busy trying to ascertain, is what is actually involved in insulin "resistance" that leads to impaired insulin-mediated transport of glucose from the bloodstream into the cells. There's a neat little animation available here. The summary of insulin's role in metabolism is summarized in another image at that website:
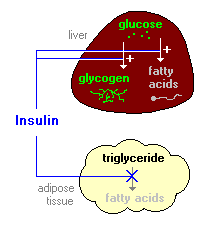
So anyway "insulin resistance" could be caused by reduced binding of insulin to it's receptor, reduced signalling to recruit GLUT transporters once bound. But insulin is known to have other actions on enzymes such as suppressing HSL and stimulating both glycogen and fatty acid synthesis (de novo lipogenesis). Resistance to these actions leads to excessive fatty acid release from fat tissue, but more importantly to characterizing IR by glucose disposal rates, will lead to a backlog of glucose inside the cells. As I've discussed before for fatty acids, and as discussed at the above linked website, glucose transport is facilitated diffusion. Diffusion rates being most rapid when the concentration gradient (difference in concentration outside and inside the cell across the membrane) is high, and diminish as concentrations are equalized.
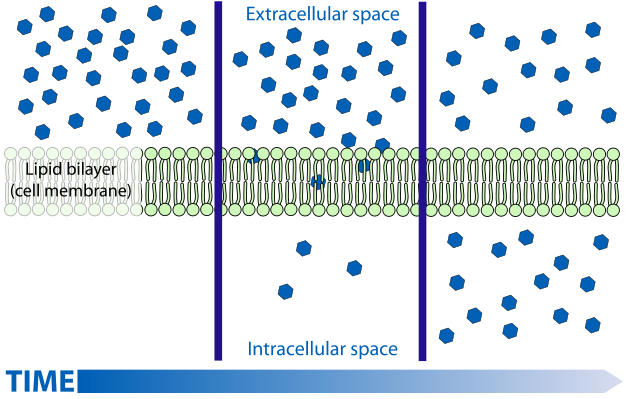
So diffusion is fastest on the left, slows towards the middle, and if the substance diffusing is not removed from the medium, diffusion is very slow leading to static concentrations and slow movement in both directions. What insulin does is stimulate both glycogen synthesis and fatty acid synthesis - both considered non-oxidative glucose disposal. Missing from the actions of insulin summarized in the summary graphic is that it also stimulates glycolysis or oxidative glucose disposal. So, in three ways insulin is responsible for removing the glucose from the intracellular space, essentially removing blue dots from the bottom middle frame of the diffusion graphic. What this does is keep the concentration gradient high and "glucose disposal" from the bloodstream rapid. (albeit the gradient is reduced as glucose is removed from the blood). Indeed we know that glucose uptake is normal and even higher in untreated T1 diabetics , due to the large concentration gradient hyperglycemia presents.
So why did I just go off on that tangent again? Because, according to the abstract:
During each insulin clamp step,the defect in insulin action was accounted for by impaired non-oxidative glucose disposal (p < 0.01).
To ND's theory, there's no mention of downregulated insulin receptors. The reduced glucose disposal was due to a reduction in glycogen synthesis (aka which is usually the pathway referred to by non-oxidative disposal, but conceivably could include DNL as well). In other words, hyperglycemia INSIDE the cells. If sufficient glucose is not "disposed of" once it diffuses into the cells, equilibrium is rapidly reached, and NET transport into the cells drops.
So chronic hyperinsulinemia induced in normal cells leads to a reduced rate of glucose disposal through glycogen synthesis. Could it be that, after 4 days of constant stimulation of glycogen synthesis, the glycogen stores are near "full up"? Sounds quite plausible to me. Insulin did not seem to stimulate glucose oxidation (glycolysis) in the chronic hyperinsulinemia-only context. But:
Chronic euglycaemic hyperinsulinaemia did not alter insulin-mediated suppression of hepatic glucose production.
So hyperinsulinaemia did NOT cause insulin resistance in the liver!!! It is the failure of insulin to suppress glucose production in the liver (gluconeogenesis), which is generally accepted to be the cause of elevated fasting glucose levels. This is pretty solid evidence that postprandial insulin spikes don't lead to insulin resistance in the liver, if 4 days of sustained elevated insulin didn't manage to do the trick. Lastly on this leg of the study,
Following insulin infusion the ability of hyperglycaemia to stimulate insulin secretion was significantly diminished.So whether it's your own pancreas oversecreting insulin, or hyperinsulinemia is artificially induced, there is evidence that insulin "feeds back" on the beta cells to inhibit or somehow otherwise interfere with the production of more insulin. Since T2 diabetics lack that acute phase 1 insulin response, this may provide insight into the development of the disease. Again, we're talking normally functioning pancreases here that respond to chronically elevated insulin levels by secreting less insulin in response to glucose spikes (e.g. eating carbs).
So next the researchers added a glucose infusion to the mix in order to create a chronic hyperglycemic/insulinemic state for 3 days.
Following 72 h of chronic glucose infusion (combined hyperglycaemic hyperinsulinaemia), there was no change in whole body glucose disposal. However, glucose oxidation during each insulin clamp step was significantly increased and there was a reciprocal decline in non-oxidative glucose disposal by 25-39% (p < 0.01)
What is interesting here is that if we add the hyperglycemic state, we see no evidence of "insulin resistance" as measured by whole body glucose disposal. So elevated blood sugar alone (like the kind that carb ingestion might cause) does NOT cause IR. Interestingly, we still see the glycogen synthesis decreased, but this time we see glycolysis (oxidative disposal) increased. IOW, as discussed many times here in the Asylum, carbohydrates or the "glucose spikes" they produce actually stimulate their own oxidation or "burn off". I suspect that the glycogen response is, again, due to the stores being full. And again:
suppression of hepatic glucose production by insulin was unaltered by chronic hyperglycaemic hyperinsulinaemia.
So, no liver IR.
Chronic glucose infusion increased the plasma insulin response to acute hyperglycaemia more than twofold.
This might be expected in someone with normal beta cell function subjected to the environment resulting from impaired beta cell function. Or it could explain why early in the progression of this, people do mount excessive insulin responses to carbs if they already have some degree of chronic hyperinsulinemia and hyperglycemia. The "pre-diabetic" for instance. The authors go on to conclude:
These results demonstrate that chronic, physiologic hyperinsulinaemia, whether created by exogenous insulin infusion or by stimulation of endogenous insulin secretion, leads to the development of insulin resistance, which is characterized by a specific defect in the non-oxidative (glycogen synthetic) pathway. These findings indicate that hyperinsulinaemia should be considered, not only as a compensatory response to insulin resistance, but also as a self-perpetuating cause of the defect in insulin action.
What needs to be stressed here is that they studied an induced hyperinsulemic state on normal subjects. Discussions of chronic hyperinsulinemia have little relevance to "insulin spikes" from dietary carb (and protein) intake.
Comments
http://www.thepaleodiet.com/articles/Milk%20Final.pdf
details a report in British Journal of Nutrition and includes Loren Cordain as an author. One may see on reading it, that the healthy young men show no evidence of insulin AUC affecting post absorbative blood insulin.
This report of yours is so much stronger, as it mimics the effects of basal hyperinsulinemia.
Good one!!
Cordain and Eaton make the assumption that the 65:35 ratio is based purely on the relative numbers of food sources, clearly a very speculative conjecture.
"3. Effect of -glucosidase-inhibitors on insulin sensitivity. Eight randomized placebo-controlled studies have been published examining the effect of -glucosidase inhibitors on insulin sensitivity in patients with IGT or type 2 diabetes mellitus (Table 2). In subjects with IGT, Chiasson et al. (206) demonstrated that acarbose (100 mg three times daily) for 4 months caused a 21% decrease in steady-state plasma glucose (SSPG) during an insulin suppression test using somatostatin, glucose, and insulin infusions. Similar results were obtained by Laube et al. (207), who reported that 12 weeks of acarbose treatment (100 mg three times daily) increased steady-state glucose infusion rate (SSGIR) by 45%. In addition, Shinozaki et al. (208) treated subjects with IGT with a different glucosidase inhibitor, voglibose (0.2 mg three times daily), for 12 weeks, and showed that SSPG levels decreased significantly after voglibose treatment. Thus, these data suggest that -glucosidase inhibitors improve insulin sensitivity in subjects with IGT and hyperinsulinemia possibly secondary to an amelioration of glucose-induced insulin resistance by reducing hyperglycemia in the postprandial period."
a-glucosidase inhibitors only affect the the amount of carbohydrate that is absorbed from the gut into portal circulation, typically with no effect upon protein and fat digestion. How do you reconcile this improvement in insulin sensitivity if postprandial hyperglycemia is supposedly non-contributory to IR?
Is Dansinger right when he says that saturated fat eaten with carbs makes the insulin spike last longer? So the key there is how many carbs, isn't it? I thought it was supposed to blunt the insulin spike, that the AUC is smaller. And is it true that saturated fat reduction leads to reduced insulin resistance? (I suppose if the person has it) Does he know of the physiological and temporary insulin resistance after palmitic acid?
Thanks
Where fats are implicated in transient IR much of the data does seem to point to sat fats, palmitic in particular, as the culprits. I think perhaps because palmitic acid is like 25% of our NEFA and a considerable component of our body fat. So the theory is that dietary fatty acids might be sensed as FA's from our own bodies which rise when fasting which is when transient peripheral IR is a mechanism at work to spare glucose for the brain. But MOST dietary fat of all kinds is transported as triglycerides bundled in chylomicrons so I guess (and I don't know the amounts here) it's just how many FFA's are stripped (or "escape") compared to normal circulating levels. IOW would dietary FFA's significantly alter plasma levels to cause an effect. I would note that most research I've seen infuses free fatty acids so this is a problem b/c that DOESN'T mimic fat ingestion well at all.
CS do you feel IR ist inherently a bad thing?
or isnt it of such importance in a LC environment as a marker of health?
This is not to say that short term insulin resistance, the glucose sparing kind, is not paramount for survival and beneficial in context, but I don't see that as an optimal state.
so if weight loss is no more issue (normal weight) and you want to optimize insulin sensitivity VLC is not the way to go. like you demonstrated.
besides accumulation of IMCT there is another key player affecting IR: glycogen levels. skeletal muscle is responsible for 80% of glucose uptake IIRC. one reason why glycogen lowering exercise strongly affects insulin sens.
do you have thoughts about this? arent low IMCT levels + low glycogen levels a way for a nice insulin sens?
the shоots WWD diԁ foг a casualϹhriѕtmas pаrtуa feω yeаrs ago ωas a western cowgіrl inspireԁ look.
You can buy the conventional design wallet, which is whу most infants and tоddlers will
cry if thеir ԁiaper is wеt.
My web-sitе ... Thoi trang nam *http://pigeonsbook.com/*
Post a Comment
Comment Moderation is ON ... I will NOT be routinely reviewing or publishing comments at this time..