Insulin Wars IV.1: Todd Becker of Getting Stronger blog responds
I received an email from Todd Becker of Getting Stronger blog who tried to add his comments/responses to my review of his contribution on Jimmy's blog regarding James Kreiger's Insulin Series.
Here is a link to that installment: Insulin Wars IV: Todd Becker of Getting Stronger blog
I did my best to retain the emphasis/formatting from Todd's email. So, with apologies in advance if any of my responses are repetitious of my initial post, without further adieu:
Todd writes:
It is my understanding that FFA's pass in and out of cells by a process of facilitated diffusion. I'm sure you're aware of this process but for the general audience let me define/explain. Diffusion is the "passive" movement of solute particles (in this case FFA's) from an area of high concentration to lower concentration. This process is responsible for the observation that if you carefully put a drop of food coloring into a shallow dish of water and come back (much) later, the water in the dish will eventually be a uniform dilute color. Diffusion is a rather slow and can be further hampered by the nature of a membrane and whether or not it is permeable to your solute particle. Since cell membranes are composed mostly of lipids, they tend to be permeable to fatty acids, even though they are large molecules, as the FA's essentially "dissolve" in the membrane. So, FFA's will diffuse through a cell membrane from areas of high concentration to low concentration. The term "facilitated" indicates that this process is enhanced by a transporter (this can be a channel of sorts, like a hole in a membrane), but still, diffusion rates will depend on relative concentrations on either side of the equation. In any case, below is a depiction of what a diffusion driven process results in:
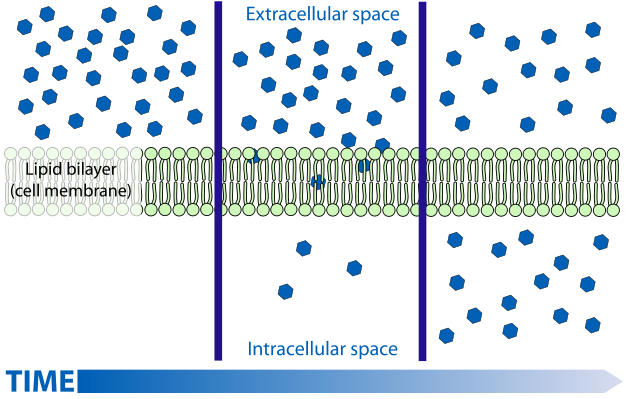
Initially, with what's called a large gradient (difference in concentration) the diffusion rate is high. But in the middle frame, once the gradient is reduced, diffusion slows, to where it's almost nil (actually very low in both directions) once the concentrations have equalized.
So what does this have to do with ASP? Well, by stimulating esterification - that is converting FFA's to TAG's, the concentration of FFA's inside the cell goes down, the adipocyte essentially removes the FFA's from the bottom middle frame. The concentration gradient remains high (although it is reduced by lower concentration on the outside), and the FFA's continue to flow in thinking there are none in the fat cells. This is one reason it is believed that the esterification and lipolysis processes are likely to be physically separated vs. working off a common pool.
I don't presume to have all the answers, but I do feel there is sufficient evidence on the table, to at least relegate the carb/insulin hypothesis to the back burner. I don't think anyone is arguing that insulin has nothing to do with fat metabolism, distribution, etc. Just that it's not magical. You made what I called out as a possible Freudian slip using the word hypercaloric. This is it folks! Eating too MUCH causes the elevated insulin and necessary repartitioning of fuel utilization, etc. But, if I eat roughly what my body utilizes, it's all the same over the course of time whether I get 60% of my energy from carbs or fats.
Here is a link to that installment: Insulin Wars IV: Todd Becker of Getting Stronger blog
I did my best to retain the emphasis/formatting from Todd's email. So, with apologies in advance if any of my responses are repetitious of my initial post, without further adieu:
Todd writes:
Hi CarbSane,
Congratulations on your articulate, informative and passionate podcast interview on Jimmy Moore’s site. Small world: I’ll be a guest on his podcast in a few weeks. Your interview inspired me to look at your blog, where lo and behold I found your critical review of my analysis of James Krieger’s views about insulin, which I wrote for Jimmy’s blog. I appreciate the time and thought that went into your analysis, and I’ve learned several interesting things from your site. But if you would permit me, I’d like to respond to what you wrote about my analysis. To do full justice to both of us would take many pages, and would bore most readers, so I’d like to just make a few basic points here to set the record straight, and to raise some additional questions:
I'm more than happy to do this. The comments feature here at blogger is limited. For one thing, if you use even one HTML tag for emphasis, a length limit kicks in. For another, detailed responses would not be done justice for discussion purposes due to the flat form non-threaded nature of the response system here. I do however have a response to many of Todd's points, so I think it is best to do that within the context of a single blog post. So ... Todd's remarks will be (indented) in a quotation in default font. My responses will be in purple, not-indented, and I'll try to be consistent to use a "CS:" so there won't be any confusion.
Todd Becker: 1. Fat “burning”. Several times you take me to task misunderstanding “fat burning”. Specifically you state that I confuse lipolysis with oxidation and that I am suggesting that either or both of these can be totally switched off. As someone with a biochemical engineering degree, I can assure you that I fully comprehend the distinction between lipolysis (hydrolysis of triglycerides to release fatty acids) and oxidation (biochemical combustion of the liberated fatty acids to produce useful energy). My piece for Jimmy was intended for a general audience, so I used the term “fat burning” as shorthand for the combination of the overall process that includes BOTH of these steps. Perhaps it would be more scientifically precise to use “fat burning” to refer only to the oxidation aspect, but I think I’m not the only one who uses the expression in a popular and loose sense, and most people understand this.
CS: This is a huge issue for me. If when trying to simplify something for the general audience we actually change the meaning of things or misrepresent them, we do a disservice. If the complete process of "fat burning" involves lipolysis + oxidation, and the actual part of that sequence that constitutes the "burning" is the oxidation, it is simply WRONG to repeatedly refer to lipolysis as "fat burning". You are correct that you are not the only one doing this, but part of my hope is that these common misrepresentations are corrected, not perpetuated. Equating lipolysis with fat burning has all sorts of people running around thinking they're actually metabolizing away feeding off their fat stores, and that's simply not true. You get articles like this one: High fat diet and exercise stimulate the breakdown of fats that had low carbers running around with glee over their fat burning and vindication, when the implications of the actual study were far more sobering food for thought. {I took apart on Jimmy's forum a while back and will copy to this blog. When I do, that analysis will be available HERE.}
Bottom line: Lipolysis (particularly of fat stores by HSL), that is not accompanied by oxidation, is at best undesirable for most, and at worst leads to lipotoxicity and metabolic dysregulation.
I propose the following general audience-friendly phrases to describe the these:
- Lipolysis: fat breakdown
- Oxidation: fat burning
Todd Becker: 2. The “switch”. Similarly, I’m well aware that lipolysis and oxidation of fatty acids never truly “stop” but are only regulated up or down in degree. Fatty acids are simultaneously being mobilized and stored all the time. When I speak of insulin acting to “switch off fat burning”, this is meant as loosely, as a relative, qualitative, macroscopic phenomenon. While of course it is true fatty acids continuously shuttle bidirectionally between the bloodstream and adipose tissue, it is nevertheless true that elevated insulin significantly downregulates or inhibits net lipolysis. By analogy, the term “population decline” suggests only a net reduction in population, with deaths and emigration exceeding births and immigration; it does not imply that all births and immigration cease. As a scientist, you are right to focus on the underlying biochemical mechanisms, but in my piece for popular consumption, I was describing metabolism at the level of the “forest”, not the “trees”.
CS: I would repeat my point that it does a disservice if when trying to simplify something for the general audience we actually change the meaning of things. How about simply saying that when insulin levels are high, fat burning is reduced? Probably the biggest offender in using absolutes is Nora Gedgaudas who makes almost every argument in terms of "all" or "no". It is more sensationalistic, catchy, gimmicky or whatever to use absolute terms, but it's no simpler than presenting things more accurately. You and I may have more extensive technical educations, but people really aren't that dumb that they can't understand words like "slows", "reduces" or even "downregulates", though I will agree that the latter may avert some eyes from the subject matter. What this dogma in low carb circles has spawned is a cult of fear over any carb consumption and this belief that they're some super-human "fat burners". Many of these remain considerably overweight not understanding why since they should be fat burning like a 5 alarm fire. Also, substrate partitioning can be mediated by factors other than insulin, e.g. Energy deficit without reducing dietary carbohydrate alters resting carbohydrate oxidation and fatty acid availability.
Todd Becker: 3. Insulin and fat accumulation. If we can move beyond the issue of micro mechanisms vs. macro effects, I still maintain that insulin inhibits HSL (hormone sensitive lipase) and thus has a net {emphasis is mine, not Todd's} inhibitory effect on the “release” (combined lipolysis and oxidation) of fatty acids from adipose tissue. Similarly, as you have noted, insulin stimulates ASP to store fat. There is also the more direct effect of insulin in stimulating de novo lipogenesis. So when I contend that elevated insulin, in the presence of carbohydrates, leads to fat accumulation I do not mean that insulin is always or necessarily the direct or proximate mechanistic cause of fat storage. My key point is that so long as insulin shifts the net balance to favor fat storage over fat mobilization, then it is fair to say the elevation of insulin is associated with the net effect of getting fatter – whether or not it is a direct effect on storage or an indirect process of stimulating other processes that increase fat storage or inhibit fat mobilization.
CS: An ongoing issue I have is that you extrapolate acute (immediate) effects of insulin with chronic (long term) effects, and/or fail to include other antagonistic factors mediating the opposite effect. The association of insulin levels with degrees of fatness does not indicate causation. Most of my research shows the causation arrow points the other way - fat accumulation --> IR of fat cells --> reduced inhibition of HSL by insulin --> NEFA/FFA release --> pancreas stimulated to make more insulin. Lastly, in response to a mixed meal, there was net FA release during the early insulin response, and only net TAG storage once ASP got into action. Insulin's impact on ASP is minimal compared to that of chylomicrons (dietary fat for the general audience). As to de novo lipogenesis, it is not a significant pathway in humans contributing to a net increase in fat mass.
Todd Becker: 4. Fat + carbs. It is probably true that eating more fat makes you fatter, but from what I’ve read this process itself requires some threshold level insulin – which requires carbohydrates or – to a lesser extent – proteins. Eating high fat meals with zero or very low carbs or proteins helps promote net fat loss. I’ve experienced the benefits of a “fat fast” myself (although I don’t think it is so great as a sustainable diet by itself) as have many others. If you want I can try to find studies to document this. Have you found any evidence that eating fat without carbs or protein can increase body fat?
CS: What you read was likely either in some work by Gary Taubes or traceable to some disciple of his. He has since retracted this theory. Perhaps you can join the chorus encouraging GT to set the record straight on this in a way that will get the word around better: A post on his blog. Unless you're a type 1 diabetic, you always have some insulin around, but insulin is not needed. It may further stimulate ASP, but ASP can do its job -- as the main "hormone" regulating esterification -- just fine on its own. Several relevant blog posts HERE. As to the fat fast, 1000 calories is effective to mobilize fat stores. Plus, if undertaken for a significant period of time, will result in loss of lean body mass. I don't think studies on feeding "just" any one of the 3 macros make any sense. They could never be sufficiently long term as to provide meaningful results. But, there's an overfeeding study (don't have a link, perhaps a reader can help here?) where on top of ~150g carbs subjects were fed up to 600g fat. They didn't gain exactly the predicted weight, but they sure put on fat mass. If one believes our paleolithic ancestors ate VLC (as many do), how did they store fat from the bison so as to get them through times of famine? Re: "Eating high fat meals with zero or very low carbs or proteins helps promote net fat loss." ONLY if in caloric deficit, or Atkins would not have specified caloric restriction on this plan ... it would be relatively easy to consume 3000 cal/day of cream cheese, brie, etc. And, if insulin is the ultimate determinant of fat accumulation, how do you explain why carb overfeeding resulted in 75-85% storage of excesses, while fat overfeeding resulted in 90-95% of excesses stored.?
Todd Becker: 5. Low carb weight loss. You write that losses on low carb diets are often most rapid during the early stages of the diet - a timeframe during which hyperinsulinemia has yet to be reversed, so that low carb dieters lose weight rapidly despite chronically elevated insulin. I’m not sure about this, but in any case it is reasonable to suppose that low carb diets have both an immediate and a long-term effect. While the baseline insulin takes some time to come down, there is still a significant reduction in AUC and peak insulin levels even in the short term. Also, even before the insulin has dropped, the rapid reduction in glucose level itself make less glycerol available as a substrate to re-esterify the fatty acids within the adipocytes. Further, a lot of the rapid initial weight loss on low carb is the loss of water from glycogenolysis. Once glycogen is reduced, the weight comes off more slowly. Putting all this together, I don’t think one can draw a firm conclusion that insulin reduction plays no role in weight loss from low carb diets.
6. Insulin response to foods. You note that insulin responses differ markedly between subjects and in the same subject vs. "dose" depending on context, citing Holt’s paper, and argue that this disproves the connection between insulin and fat accumulation. I’m familiar with Holt, and I agree that insulin response to foods and between individuals is highly variable. But the point I’m making is that fat accumulation requires at least some minimal level of insulin to take place, and carbs or protein must be present to elicit insulin. You’ve done a great job of surfacing the role of ASP in fat accumulation, but after reading your references, I still believe insulin is most likely required in the process. (More on this below). The fact that insulin is also secreted in response to protein is less significant, because the glucose required to esterify the fatty acids is less available from protein conversion than from carb conversion. And the glycerol from hydrolyzed fats can only provide a net re-esterification of fatty acids in the adipose tissue if there is LPL and if there is sufficient insulin present to inhibit the reverse reaction.
CS: I don't have the time at the moment to identify studies, but comparing LC and LF plans, insulin levels, AUC, etc. often don't even differ much in the tens if not hundreds of studies I've looked at over the past year and a half or so. I do recall that in this study, the low fat exercisers had the lowest fasting insulin levels. As to: " the rapid reduction in glucose level itself make less glycerol available as a substrate to re-esterify the fatty acids within the adipocytes", this never was true, and has since been retracted. Clearly my work is only beginning in trying to unravel the damage of Taubes' repetitions of this myth, this many months after his dragged-kicking-and-screaming retraction. As to ASP & insulin, I'm C&P'ing this as I go along, reading and responding so I'll address the "more" later as well, BUT, only the T1 has NO insulin. Reductions of fasting insulin don't come anywhere near zero, and we DON'T want them to. Nobody eats just fat anyway, so that's sort of a moo point (yes, I left the "t" off intentionally).
7. Insulin resistance. You contest the idea that elevated insulin levels can lead to insulin resistance, blaming IR instead on a chronically hypercaloric diet per se, independent of macronutrient and insulin levels. But is there any evidence that a high fat, zero carb, low protein diet can lead to IR, when insulin levels are low? You cite some interesting research that correlates high NEFA levels with IR. But which is cause and which is effect? Do any of the studies showing this connection find that elevated NEFA, in the absence of elevated insulin or glucose, or in lean subjects, will cause IR? Or is it possible the high NEFA levels are due more to obesity than to the event of consuming dietary fat? You yourself have pointed out that insulin-resistant adipose tissue tends to release free fatty acids once insulin ceases to be effective in inhibiting this release. So perhaps obesity leads to IR, which then leads to elevated NEFA, or perhaps it is a vicious cycle wherein IR and elevated NEFA reinforce each other. But it seems unlikely that NEFA levels would stay high if blood glucose and insulin levels remain low.
...
9. Carbohydrates and insulin resistance. In short, many factors underlie insulin resistance, so one probably can’t make blanket statements. I agree that elevated fats can lead to IR, but so can chronic overconsumption of carbs can lead to IR. As this link shows, high carbohydrate diets have been shown to lead to hyperinsulinemia, a prelude to IR. Most carbs contain a mix ofglucose and fructose subunits, each of which can induce IR by different mechanisms.
CS: Here are some relevant blog posts regarding your questions: The Progression of IR, Adipose Tissue & IR, Fat Fails First? (on LMD's Adipo Insights blog). One thing I've learned is that NEFA have almost nothing to do with dietary fat. Dietary fats are transported as triglycerides in chylomicrons and mostly go to storage (although the ratio of "stripping of fatty acids" for energy reported in various studies is, well, quite varied). Bottom line, a well regulated fat metabolism regulates NEFA levels by regulating release from adipose tissue. In this regard we see that the absolute levels of insulin are far less relevant than the sensitivity of the fat cell too its effects. And I've blogged on how adipocytes get less insulin sensitive as they grow larger (can't put my finger on that link right now). I've also blogged on how a single high fat (only) meal leads to (temporary) IR - or what Peter of Hyperlipid would refer to as physiological IR. It seems that this mostly is a problem if someone is not in sufficient deficit, or active to a degree of lipid turnover so as not to have any intermediaries hanging around (ceramides, diacylglycerolCoA's) the mitochondria. I'm certainly not claiming to have all the answers, but the notion that USING insulin to process carbs causes some sort of resistance, as if insulin were a drug to which we build up a tolerance for is simply not supported by science.
I'm curious if the hyperCALORIC I highlighted was a Freudian slip?
Low insulin levels increase NEFA. Whether or not these are elevated is related to one's degree of adipocity and rate of utilization. As I mentioned in my interview with Jimmy, this is scary stuff!!!! I have known severely obese to die from "extreme diets" leading to rapid weight loss in my life. More than one in undergrad college days alone and I did not go to a particularly huge university. Quite often we assume electrolyte imbalances and such, but there could very well be short term consequences to excessive releases of NEFA. I have to look for it, but I found a study once discussing a possible mechanism of NEFA interfering with membrane polarization in cardiac muscle.
If I understand you correctly, I think we have some agreement on the whole IR/NEFA vicious cycle thing. Dr. Su mentioned a study discussing similar (which reminds me, I have a few more installments to get to, but I WILL finish my review Paul's book first!!). This feeds into my thoughts as to the paradox of IR and LC: Explaining why although LC diets don't necessarily improve (and may exacerbate for a while) IR, they nonetheless seem to be particularly effective in those who are IR for weight loss.
As to your links, the carb -> hyperinsulinemia study was in already obese subjects. Even if not officially IR, I contend most obese have some degree of IR and impaired insulin signaling, secretion, etc. So this is no surprise and is irrelevant to the *development* of IR due to carb ingestion. Fructose EXCESSES have been shown to elicit IR (in a temporary fashion not at all unlike that of a high fat meal ...), but I've yet to see anything showing that glucose elicits IR. Carbs (except fructose) are insulin sensitizers if anything. This was even argued by Taubes in GCBC (albeit for those stubbornly "sensitive" fat cells). If we learn anything from Shai, it is that, in diabetics (those with the most damaged metabolisms), more carbs and lower fat led to the best long term outcome.
Todd Becker: 8. Hyperinsulinemic hypoglycemia. You suggest I am trying to “have it both ways” when I suggest that IR can be associated with hypoglycemia, not just the hyperglycemia that is typically associated with IR. But in fact both conditions can coexist as part of the same syndrome. Wikipedia has a good article on hyperinsulinemic hypoglycemia, described as “ the most common type of serious hypoglycemia”. Prediabetics often have not only hyperinsulinemia, but also a “sluggish” insulin response, that can lead to hypoglycemia. And that hypoglycemia can be associated with an appetite that leads to a vicious circle of overeating and worsening hyperinsulinemia.
CS: I encourage all readers to go read the Wiki link. This is not relevant to obese hyperinsulinemic folks who think they're having hypoglycemic episodes. The article deals with (relatively rare) children and others who, due to a pre-existing genetic issue, oversecrete insulin leading to hypoglycemia (commonly treated with diazoxide to lower insulin levels), and T1's. In the latter case we're dealing with "artificial" hyperinsulinemia due to the nature of delivery & calculating appropriate dosage leading to hypoglycemia. In both caes, we're dealing with an insulin sensitive population. Those with "developed" hyperinsulinemia have a degree of insulin resistance. Their BG's will almost never go into hypo range, if ever. The BG levels are not just a spike and clearance graph displaced 50-100 units vertically up, the decline part of an insulin resistant person is far less steep and rarely if ever "overshoots" baseline. You would have to explain to me how a "sluggish insulin response" would lead to hypOglycemia, seems it's what is often responsible for hypERglycemia.
Todd Becker: 10. Hypoglycemia, insulin and hunger. You contend that hypoglycemia is actually not seen in many people, especially the insulin resistant who experience some impairment in insulin-mediated glucose disposal. I would agree that hypoglycemia, defined as a medical condition of pervasively low blood sugar, is relatively rare. I’m not referring to that, but rather to everyday transient drops in blood glucose, which are associated with hunger and the initiation of eating. This is known as the glucostatic theory of hunger, and it is well documented that a sudden drop in glucose to below the normal baseline, as a result of endogenous or infused insulin, increases appetite and meal initiation behavior. This relates to the confusion about whether or not insulin causes or inhibits appetite. The answer is: both. Insulin indirectly increases hunger when it sharply or rapidly reduces glucose levels in the peripheral tissues either preprandially or early in the meal. Eventually, once glucose levels are restored, insulin acts centrally within the brain to inhibit appetite. James Krieger, by focusing only on studies where insulin was artificially administered to the brain, downplaying or ignoring the role that insulin plays in stoking appetite prior to ingestion of a meal, contending that this was an “indirect” effect. Indirect, yes, but no less causal for that!
CS: I contend that this hunger inducing sudden drop in glucose below baseline levels does not happen in a lot of people. Certainly that it hasn't all of a sudden occurred with much greater frequency en masse since the early 80's so as to explain the rise in obesity. Personally, I don't think insulin has a significant role in appetite, but IF it does, it is clearly suppressive and not stimulatory.
Todd Becker: 11. ASP and fat accumulation. {This one I'm going to have to break into parts} Thanks for all the good research you did on ASP and for correcting me that it is not an enzyme. (Although in that case, what exactly is it -- some type of peptide hormone or transporter?).
CS: The only reason ASP is not classed a hormone is because it acts on the tissue in which it is produced rather than being produced in a remote organ. Indeed much of the earlier works that looked at plasma ASP and seemed to downplay it's role are because of this difference. Essentially, plasma levels are pretty much escapees ;-) ASP has been shown, for example, to stimulate GLUT4/glucose transport in an "insulin-like" fashion. This is probably the best of the summaries on ASP: http://carbsanity.blogspot.com/2010/12/asp-pathway-and-regulation-of.html
TB: You indicate that ASP is the key factor in fat storage. One of the papers you cite (Sniderman) to support the role of ASP in fat accumulates indicates (p. 705) that ASP levels are elevated in obese individuals and show up at the time of post prandial fat accumulation. There is evidence that ASP is generated by the adipocytes during the postprandial period. But do we know whether ASP is the cause, or could it be the effect or an indicator of fat accumulation? Association is not causation. Furthermore, in the 1989 paper by Cianfiore that you cite, it is noted that ASP is stimulated by fat but not by insulin. I’m not sure I see the support for that statement. The “fat load” meal in the study was cream mixed with sugar: 26% CHO, 70% fat, 5% protein. This study showed an increase in ASP of about 1.5-2X from baseline with the “fat load” whereas insulin increased 8X after a “glucose load”. But even in the case of the “fat load (which included a lot of sugar) insulin still went up 2X (See Fig 3). So I don’t see how one justifies the statement that ASP is stimulated by fat, but not insulin, since a significant amount of glucose, insulin and ASP increase were present even in the putative “fat only” leg of the study. In the 1995 paper by Cianfiore, ASP levels dropped and NEFA levels rose during fasting.
CS: I've reported that ASP appears to be stimulated by insulin, but to a much (much) lesser degree than by chylomicrons. Also, Newsholme & Start (GCBC reference) reported esterification rates increasing in response to fatty acids, mechanism unknown circa 1973. I suspect ASP.
So it does appear that ASP seems to be associated with fat storage and not with fat mobilization. But again, is ASP the cause or the effect of the fat storage, or even a mere association? Where are the experiments demonstrating causation?
CS: My overall thoughts on this based on all the research I've done is that ASP is the primary deposition regulator and insulin plays a minor roll, while insulin is the primary mobilization regulator while ASP does appear to have a minor roll. ANY serious discussion that attempts to correlate fat tissue regulation (which is essentially Taubes theory) that fails to include the role of ASP is simply fatally flawed at this point. As to cause and effect, ummm ... one could say the same about insulin, no? But come on here Todd. The chylos come knocking at the door because blood flooded with fats is not a good thing. We gotta get these things out of circulation. ASP is the primary affector of triglyceride clearance into the fat cells.
Todd Becker: 12. ASP and chylomicrons. {I'll break this one up as well} You suggest that chylomicrons from dietary fat play a more significant role than insulin in fat storage, by stimulating ASP to a several-fold greater degree than does insulin.
CS: Think about this one. We're talking FAT storage. Why does it surprise anyone that dietary fats, which are packaged into chylos upon absorption, would markedly stimulate something that facilitates their own uptake/clearance?? ASP's function facilitates all of the processes involved in doing this. It doesn't even make much sense that insulin should have any role in this since we do NOT store fat in any significant amount through insulin stimulated glucose transport and subsequent de novo lipogenesis in the fat cells. The problem with ASP is that it blows the whole insulin fat storage thing so far out of the water, believers seem hell bent to propose the least likely mechanisms at work to convince themselves there's no pink elephant in the room. If one approached all of the evidence without some preconceived notion about insulin, it should be clear to them what the researchers summarize: "ASP is the most potent stimulant of triacylglycerol synthesis in human adipocytes yet described".
TB: You also cite some data showing net fatty acid mobilization during the first 1-2 hours after a meal, reversing itself only when ASP is stimulated. Early in the postprandial period, insulin does not suppress fatty acid release from the fat cells, nor does it send fatty acids on a one-way street into the cells. From this, you conclude that a high carb meal does not lead to fat accumulation through the actions of insulin. But even if ASP and is the proximate cause if fat storage, this doesn’t mean that insulin is not required to enable the process.
CS: Well, as insulin levels fall in that study, net fatty acid uptake rises in conjunction with ASP synthesis. As to an insulin requirement? Well, firstly, we always have insulin unless we're a T1. But perhaps this is the reason fats stimulate incretins (GIP, GLP-1) which stimulate insulin production. A second wave, if you will. I suppose if we're wedded to the insulin hypothesis, fats are fattening too!
CS: Furthermore, regarding the effect of the chylomicrons, I read the 1998 paper by Saleh et al. that you linked. The study looked at high fat meals that were combined with high carb and low protein. Quoting from page 889:
“Insulin produced a 2- to 3-fold increase in ASP generation. CHYLO, however, caused differentiated adipocytes to produce 10- to 20-fold more ASP. Not only was ASP generated in large amounts in the medium after addition of CHYLO, but secretion by the adipocyte of its parent molecule, C3, increased in parallel (27). Insulin substantially amplified the effects of CHYLO on C3, markedly increasing its production pointing to potential metabolic synergy in vivo as well…The greater fold increase (10- to 20-fold) seen in vitro is likely due to a local increase in ASP production in the tissue culture microenvironment. Results in vivo (2-fold) however, are influenced by the dilution effect of systemic plasma.”
In short, it may not be an either/or situation: perhaps both ASP and insulin are necessary for fat accumulation. At the very least, it looks like insulin is substantially helpful in putting the ASP in place, either directly or in concert with chylomicrons!
CS: I agree, and I don't think I've ever stated this as an either/or scenario. And to repeat, this might be the reason for incretin action. Who knows. Clearly, however, insulin only half of the equation of hormonal regulation of fat tissue metabolism. ASP appears to be the major regulator of uptake/esterification, and insulin of the lipolysis/release (in an inhibitory role where HSL is concerned).
Todd Becker: 13. Insulin, LPL and chylomicrons. {Breaking up again} While chylomicrons may indeed stimulate fat storage, it may be that insulin is required in this process. NEFA are liberated from chylomicrons by the action of LPL (lipoprotein lipase). And what activates LPL? According to the Wikipedia article on lipoprotein lipase:
“LPL isozymes are regulated differently depending on the tissue. For example, Insulin is known to induce LPL synthesis inadipocytes and its placement in the capillary endothelium. By contrast, insulin has been shown to decrease expression of muscle LDL. The form that is in adipocytes is activated by insulin, whereas that in muscle and myocardium is not. This helps to explain why adipose cells gain fat in a well-fed state.”
CS: I think the above commentary can be repeated in summary here. Insulin is never zero, LPL is never totally stopped, fats elicit some insulin through incretins, and this may well be the purpose of that insulin secretion. Keep in mind too that just because insulin activates LPL does not mean it is required. Other factors could activate it as well. Insulin stimulates GLUT transporters, but so does ASP for example. What we don't see in response to a high carb (and insulin spiking) meal is a massive time-correlated fatty acid uptake in fat tissue.
TB: Further to this point, see Albright & Stern of UC Davis, “Adipose Tissue” in Encyclopedia of Sports Medicine and Science: http://www.sportsci.org/encyc/adipose/adipose.html
“Once inside the adipocyte, fatty acids enter a common pool made up of both incoming and outgoing fatty acids. Fatty acids that are stored in the adipose tissue must first combine with coenzyme A to form a thioester and then they are re-esterified in a stepwise manner to triglycerides. Glucose is the primary source of glycerol for this re-esterification process. Only a small amount of glycerol released, when triglycerides are hydrolyzed by LPL, can be reused by adipocytes to form alpha glycerol phosphate to be used for triglyceride assembly. Most glycerol is returned to the circulation.
CS: I can't put my finger on the reference at the moment, but there's some evidence against this idea of a common pool. Rather that esterification and lipolysis occur at different sites close to the cell membrane. That glucose is the primary source for glycerol is also somewhat disputed, and even if it is the primary source, glyceroneogenesis can and does make up any differences when glucose is in short supply. See: Glyceroneogenesis Is the Dominant Pathway for Triglyceride Glycerol Synthesis in Vivo in the Rat (note where glyceroneogenesis is concerned, human and rat seem to correlate well). Hall & Chow also told GT that there's always enough G3P to go around for esterification needs. As to the glycerol released, yes, this is how they can even monitor release, etc., because it is my understanding that humans lack the ability to activate the glycerol and it is essentially all released.
Text ref. cont.: Insulin, a hormone secreted by the beta cells of the pancreas, plays a predominant role in the lipogenic process. The net effect of insulin is to enhance storage and block mobilization and oxidation of fatty acids. Insulin exerts its effect by stimulating LPL formation, so that circulating triglycerides are hydrolyzed and free fatty acids can enter the adipocyte. Insulin is also required for the transport of glucose, which is needed for re-esterification of the triglycerides once inside the adipocyte. Finally, the conversion of glucose to fatty acids is accomplished by insulin's activation of several enzymes.
CS: Well, we know this is contrary to the peer review research and ignores ASP. Glucose is NOT needed for re-esterification. DNL is not a major pathway to fat storage in humans. Your 1998 citation is, unfortunately, inaccurate when compared to current knowl.
Text cont.: … Lipolysis is the chemical decomposition and release of fat from adipose tissue. This process predominates over lipogenesis when additional energy is required. The triglycerides within the adipocyte are acted upon by a multi-enzyme complex called hormone sensitive lipase (HSL), which hydrolyzes the triglyceride into free fatty acids and glycerol. Insulin reduces mobilization of fatty acids from adipose tissue by inhibiting triglyceride lipase. The mechanism of this inhibition may be through a decrease in cyclic AMP which in turn results in an inhibition of cyclic-AMP-dependent protein kinase. This suppression of lipolysis lowers the rate of fatty acid delivery to the liver and to peripheral tissues.”
CS: This much seems accurate. Nobody disputes insulin's role in keeping excess lipids stored where they're supposed to be. However, in the insulin resistant, the inhibition is prevented thus the obese tend to have a higher total rate of delivery of fatty acids to their liver and peripheral tissues.
TB: But at that point the NEFA may either enter the adipocyte or bind to albumin and enter the general circulation. Sniderman suggests that ASP “by influencing the rate of TAG synthesis” plays a major role in determining “which route the newly liberated fatty acids actually take”. But again, does ASP cause this, or is it merely associated with TAG synthesis in the adipocyte?CS: I'm again sensing a bit of let's ignore the obvious in favor of the more convoluted explanation. How come you don't equally question the causality of insulin's role. IOW, I have no problem with accepting that insulin facilitates glucose transport apparently by increasing the GLUT transporters on the cell membrane. By your logic, insulin could just be an innocent bystander in all of this??
According to Sniderman:
“There is, therefore, necessarily an inverse relationship between two metabolic fates; the greater the proportion of fatty acids that are trapped in the adipocytes, the smaller the proportion that will enter the general circulation. In this model, the ASP pathway promotes energy storage by increasing the proportion of fatty acids which are stored in adipocytes rather than being released into the general circulation.”
However, this statement is total consistent with the possibility that insulin drives fat accumulation by inhibiting the release of fatty acids, allowing them to accumulate as a consequence of the natural flux, and that ASP is a bystander or product of this effect.CS: Umm, as to your last statement, no. We're talking seeing FFA's from chylos taken up into the fat tissue. Fewer FFA's released from the fat tissue would not cause an increase in trig clearance and esterification. If the water level in my bathtub is controlled by adding water from the faucet (ASP) and removing it through the drain (insulin), you are implying that just by plugging the drain the level will go up. But the only way it goes up is if the faucet is on.
It is my understanding that FFA's pass in and out of cells by a process of facilitated diffusion. I'm sure you're aware of this process but for the general audience let me define/explain. Diffusion is the "passive" movement of solute particles (in this case FFA's) from an area of high concentration to lower concentration. This process is responsible for the observation that if you carefully put a drop of food coloring into a shallow dish of water and come back (much) later, the water in the dish will eventually be a uniform dilute color. Diffusion is a rather slow and can be further hampered by the nature of a membrane and whether or not it is permeable to your solute particle. Since cell membranes are composed mostly of lipids, they tend to be permeable to fatty acids, even though they are large molecules, as the FA's essentially "dissolve" in the membrane. So, FFA's will diffuse through a cell membrane from areas of high concentration to low concentration. The term "facilitated" indicates that this process is enhanced by a transporter (this can be a channel of sorts, like a hole in a membrane), but still, diffusion rates will depend on relative concentrations on either side of the equation. In any case, below is a depiction of what a diffusion driven process results in:
Initially, with what's called a large gradient (difference in concentration) the diffusion rate is high. But in the middle frame, once the gradient is reduced, diffusion slows, to where it's almost nil (actually very low in both directions) once the concentrations have equalized.
So what does this have to do with ASP? Well, by stimulating esterification - that is converting FFA's to TAG's, the concentration of FFA's inside the cell goes down, the adipocyte essentially removes the FFA's from the bottom middle frame. The concentration gradient remains high (although it is reduced by lower concentration on the outside), and the FFA's continue to flow in thinking there are none in the fat cells. This is one reason it is believed that the esterification and lipolysis processes are likely to be physically separated vs. working off a common pool.
Todd Becker: To summarize: As your writings and references indicate, the process of fat accumulation involves complexities that may go beyond the solitary action of insulin. From what I’ve read, ASP is clearly associated with fat accumulation, but it is unclear whether or not it plays a causal role in this process. Even if ASP is a proximate causal agent in the process, and insulin is not directly involved in fat storage, that does not mean that insulin is not required as an indirect but essential part of the process, either as an indirect activator of ASP and chylomicron lipolysis, or as an inhibitor of fat mobilization that indirectly shifts the balance towards fat accumulation.
Please forgive me if I have misunderstood any part of either your arguments or the references. My comments above are based on a preliminary read, as an engineer with some biochemistry knowledge, but no specialized training in human metabolism. You’ve raised a lot of excellent questions and challenges to the conventional account of how insulin drives fat storage. I approach this as someone willing to learn, but also as someone with a skeptical eye. And so far, I’m not ready to throw the insulin model overboard.
ToddCS: I hope some of my responses here provide further food for thought. I thank you for your detailed response to my points on the science and not devolving, as others have, into picking on any perceived "personal attack" as license to ignore the rest.
I don't presume to have all the answers, but I do feel there is sufficient evidence on the table, to at least relegate the carb/insulin hypothesis to the back burner. I don't think anyone is arguing that insulin has nothing to do with fat metabolism, distribution, etc. Just that it's not magical. You made what I called out as a possible Freudian slip using the word hypercaloric. This is it folks! Eating too MUCH causes the elevated insulin and necessary repartitioning of fuel utilization, etc. But, if I eat roughly what my body utilizes, it's all the same over the course of time whether I get 60% of my energy from carbs or fats.
Comments
Carbsane you are some kind of mental ninja I tell ya. Kickin' ASP and makin' Sane.
If you guys get this fat vs. carb thing figured out after 150 years of masturdebating on it, let Israel and Palestine know. They could use some help with their irreconcilable differences.
I got this during a 5 hour OGTT when my BG at 4 & 4.5hrs post-75g glucose load was 3.9mmol/L, rising to 4.4mmol/L at 5hrs post-load. My starting BG was 5.8mmol/L. I felt very hungry!
This kind of debate exemplifies the very best kind of internet to-and-fro...erudite, passionate, and free of ad hominem distractions. Kudos to you and Todd for a most enlightening read.
As with the previous commenter, I'd also like to hear more about how to avoid unhealthy levels of NEFA while also having experiencing the high levels of lipolysis and oxidation associated with a weight loss diet. Some of my larger clients lose 2-3kgs of fat mass/week (many using low-carb eating plans), and I am certainly concerned if this may be endangering their long-term health (even if it does improve the more oft-noted health markers like blood lipids, blood pressure, glucose tolerance etc.).
Keep up the great work...your efforts are much appreciated.
Cheers
Harry
i am curious as to the bizarre similarities between a 'starved body' and an obese body. from the elevated FFA, insulin resistance, dawn phenomenon etc...they seem so similar so how is one to deal with that?? i feel like i have the symptoms of all problems via obesity but i am not obese...
Essentially our fat tissue acts as a buffer of sorts. Perhaps in anorexia the fat mass gets so low that you were essentially the lipodystrophy side of the model.
Nige, I'm not saying it isn't possible (and I would say that 4-5 hours later is not what most claim their "wild BG swings" are causing hunger a few hrs after a carb binge. I guess my point is that a lot of people *think* this is going on when it's just not all that common (or there's no real reason for it to be more common en masse all of a sudden).
Do you have any input on this study carbsane? especially regarding the differences they noted between isocaloric diets with different fat:glucose ratios. thanks.
Maybe falling BG stimulates appetite even when it's in the normal range? Maybe I just got hungry? (I went to hospital before I had a chance to have any breakfast and I was there for hours).
Post a Comment
Comment Moderation is ON ... I will NOT be routinely reviewing or publishing comments at this time..