Adipose tissue as a buffer for daily lipid flux ~ Keith Frayn 2002
Adipose tissue as a buffer for daily lipid flux
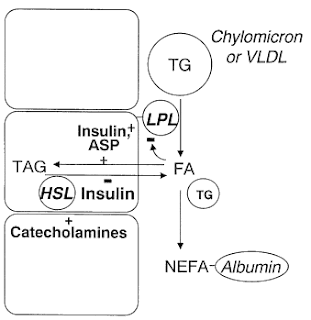

Insulin resistance occurs in obesity and Type II (noninsulin-dependent) diabetes mellitus, but it is also a prominent feature of lipodystrophy. Adipose tissue could play a crucial part in buffering the flux of fatty acids in the circulation in the postprandial period, analogous to the roles of the liver and skeletal muscle in buffering postprandial glucose fluxes. Adipose tissue provides its buffering action by suppressing the release of non-esterified fatty acids into the circulation and by increasing triacylglycerol clearance. In particular, the pathway of ‘fatty acid trapping’ (adipocyte uptake of fatty acids liberated from plasma triacylglycerol by lipoprotein lipase) could play a key part in the buffering process. If this buffering action is impaired, then extra-adipose tissues are exposed to excessive fluxes of lipid fuels and could accumulate these in the form of triacylglycerol, leading to insulin resistance. These tissues will include liver, skeletal muscle and the pancreatic beta cell, where the long term effect is to impair insulin secretion. Adipose tissue buffering of lipid fluxes is impaired in obesity through defects in the ability of adipose tissue to respond rapidly to the dynamic situation that occurs after meals. It is also impaired in lipodystrophy because there is not sufficient adipose tissue to provide the necessary buffering capacity. Thus, the phenotype, at least with regard to insulin resistance, is similar with both excess and deficiency of adipose tissue.

Below is a bullet-point paraphrased version of the figure caption in the article:
- At all times we have the Triglyceride/Fatty Acid cycle in adipose tissue where 3FA + glycerol are esterified to triglycerides and vice versa. The relative rates are controlled by insulin and ASP's actions on the enzymes involved.
- Fasting: Hormone Sensitive Lipase - HSL - acts inside the fat cells to break down triglycerides to glycerol + fatty acids - FFA/NEFA.
- This results in net outflow of FFA from adipose tissue to be delivered to other tissues for energy.
- Postprandially (after a meal), insulin activates lipoprotein lipase - LPL - outside the cells to break triglycerides in circulation down to glycerol + FFA for transport in to cells.
- Insulin stimulates the pathway of fatty acid esterification, reinforced by ASP. {I would note that this is in the context of a mixed meal, however absent significant insulin response to a high fat low carb meal, I've discussed here several studies demonstrating that the chylomicrons transporting dietary fat stimulate ASP and stimulate uptake/esterification. If ASP cannot do this w/o sufficient insulin, we have inadequate trapping of FFA's - not desired!}
- HSL is suppressed by insulin thus reducing outflow of FFA's. The combination of stimulated esterification and decreased HSL lipolysis results in a net inward flow of fatty acids from bloodstream to fat cells.
Frayn summarizes this process as: "the ‘buffering’ action of adipose tissue against lipid fluxes in the circulation." The normal time-course following a mixed meal is shown below.

This will likely get a bit repetitive, but from the article:
The major mechanisms that contribute to the buffering of fatty acid flux in the postprandial period are probably threefold and not unrelated: (i) suppression of NEFA release, (ii) increased clearance of circulating triacylglycerol, and (iii) suppression of endogenous triacylglycerol secretion. Note: triacylglycerol = triglyceride
Bullet-point paraphrasing of this section:
- The suppression of NEFA release is rapid, going from peak fasting rates to virtually zero in an hour and a half.
- NEFA release is regulated inside adipocytes
- The increased clearing of circulating triglycerides = "fat deposition" occurs more slowly following a mixed meal and peaks 3-5 hours after a meal coinciding with peak circulating triglyceride levels.
- Some of the fatty acids "liberated" by LPL are not transported/esterified, rather enter the bloodstream as NEFA.
- Dietary fats enter the plasma NEFA pool rapidly.
- The partitioning of NEFA between adipocyte uptake and circulation seems to be regulated within the adipocyte cell.
- The high fatty acid levels within the adipocyte in the fasted state, that flow out "down" a concentration gradient out into circulation, are incompatible with uptake of fatty acids against the gradient. Almost all LPL-NEFA goes into circulation initially
- As insulin clamps down on HSL, fatty acid levels inside the adipocyte fall, eventually leading to a concentration gradient favoring uptake of LPL-NEFA.
- This occurs in concert with stimulation of esterification (triglyceride formation) that further favorably impacts the concentration gradient towards uptake.
- Frayn does not go into detail but states: "Intracellular fatty acid binding proteins and acyl-CoA synthase are, of course, also involved in this process, as also are the putative fatty acid transporters" IOW, it's not all about the lipases and passive diffusion.
- This fatty acid trapping probably regulates the flux (amount and rate) of the triglyceride/fatty acid cycle. Accumulated NEFA at sites of LPL-lipolysis can lead to detachment of the enzyme from it's action site along with the remaining triglycerides in the "remnant" particle.
- This increases fatty acid delivery of triglycerides to the liver and other tissues.
- Direct Quote: "The pathway of fatty acid trapping is regulated by insulin, as noted above, and also by the acylation stimulating protein (ASP) derived from components of the alternative complement pathway produced by adipocytes."
- The partitioning of LPL-NEFA varies from almost zero in the fasted state, to around 90% within 1-2 hours after a mixed meal.
- A postprandial reduction in endogenous (by the liver) triglyceride formation is suggested - analogous to suppression of endogenous glucose production following a meal. Direct human evidence is lacking due to insufficiencies of experimental methods, but animal models support this.
- This action in the liver is "likely to be reinforced by suppression of NEFA release from adipose tissue."
- NEFA production and hepatic triglyceride secretion (VLDL) are closely related. However reduced NEFA has less of an effect on lowering VLDL production than insulin.
- Direct Quote: "It might be noted that NEFA delivery from adipose tissue is now seen also as a major determinant of hepatic glucose output, thus giving adipose tissue a role in signalling nutrient requirements to the liver."
Frayn goes on to make clear that adipose tissue is not the only tissue involved in the "buffering" of lipids as the liver is clearly involved and skeletal muscle can take up and store fatty acids. However, uptake and "fatty acid trapping" is only upregulated in the postprandial state in adipose tissue, and adipose tissue is the only tissue that releases NEFA into circulation.
"Hence, we believe that it has a special place in ‘buffering’ of lipid fluxes."
In accordance with his previous review, Adipose tissue and the insulin resistance syndrome, blogged on here, Frayn discusses IR of the fat tissue:
There is much evidence showing that the function of adipose tissue is disturbed in insulin-resistant states. The ability of insulin to suppress fatty acid release is impaired in obesity and other insulin-resistant states. ... Similarly, the ability of insulin to up-regulate LPL activity and hence triacylglycerol clearance in adipose tissue is impaired in obesity ... The pathway of fatty acid trapping in adipose tissue could also be defective in insulin resistance, thus adding to impaired buffering of fatty acid flux. These and similar studies ... show that the hyperinsulinaemia associated with obesity is not sufficient to produce normal adipose tissue metabolism in the postprandial period. These impairments in adipose tissue function will lead to less effective buffering of fatty acid fluxes in the circulation, manifest as increased postprandial NEFA and triacylglycerol concentrations.
It is increasingly recognized that insulin resistance is associated with increased triacylglycerol concentrations in insulin-responsive tissues including skeletal muscle ... and the liver. In addition, prolonged exposure of pancreatic islets to fatty acids results in impairment of insulin secretion, and (in rodents) lipid accumulation in pancreatic islets is characteristic of states associated with insulin resistance and Type II diabetes.
... These and other observations have led to the concept of ‘lipotoxicity’ in the pathogenesis of Type II diabetes. The key point of our hypothesis is that this lipotoxicity reflects impairment of the normal ability of adipose tissue to buffer excessive fatty acid flux, especially in the period following meals.
Essentially, IR of the adipocytes leads to dysregulation of this buffering action resulting in elevated NEFA in circulation. This is due to the combination of failure to clamp down on NEFA release following a meal coupled with inefficient trapping of dietary fats that have undergone lipolysis with intent to transport into fat cells. Fats not properly trapped and stored in adipose tissue or burnt for fuel are deposited somewhere: other tissues, aka "ectopic fat deposition".
In calorie deficit on a VLC/VHF diet, it would seem that (a) the dietary fat load is not especially high, and (b) it's unlikely that lipids will build up in skeletal muscle (to where intermediates like diacylglycerols and ceramides build up which seem to cause skeletal cell IR) and we're burning more fat for fuel than carb. The improvements in IR on low carb diets are far from surprising to me, even very high fat (by percent) versions of the diet. And even if superior compared to low fat diets. Why? It is not at all unusual for a low carber to spontaneously reduce intake well below the usually prescribed 500-1000 cal deficit prescribed by responsible CRD plans. To me, the jury is out, however, whether calorie deficit protects the pancreas. However the effect of NEFA on the pancreas doesn't seem to be one of damaging it (at least it seems to take quite a long time for that to occur), but rather to interfere with insulin production and secretion rates. I'll be blogging on this shortly.
But what of the long term low carber at maintenance? Particularly if that maintenance involves a reduced but still higher than optimal fat mass. If the diet does accomplish what it attempts to - lowering insulin levels - which it does indeed seem to do in some, and it does increase lipolysis and fatty acid release from fat cells as we're told is the desired outcome, is this not mimicking the metabolic milieu of the dysfunctional adipocyte? The skeletal muscle IR of the "fat burner" may well be benign, but what of the pancreas? Insulin resistance of the adipocytes leads to dysregulation of this buffering action resulting in elevated NEFA in circulation. This is due to the combination of failure to clamp down on NEFA release following a meal coupled with inefficient trapping of dietary fats that have undergone lipolysis with intent to transport into fat cells. Vascular endothelium? Liver? The fact that low carber livers pump out fewer triglycerides may well NOT be cause for celebration because that means fatty acids are finding their ways elsewhere.
I remain concerned over a diet that produces a fasting-like circulating metabolic mileu in the well-fed state.
Comments
Thanks for this post.
I'm wondering whether the elephant in the room here may be the absolute amounts of dietary fats (as opposed to the relative percentage to protein and carbs).
Seems to me that the positive effects of calorie deficits on primary health markers (even with VLC/VHF diets) may be being mediated by the modest amounts of dietary fat (in absolute terms).
Perhaps we need to start focusing more on absolute values, and less on ratios?
Cheers
Harry
No Sue, I don't think I have lipodystrophy. My thighs are hardly emaciated, just not as big as they used to be in comparison to the upper body or vice versa.
Harry: I think the followup post on Fatty Acid Trafficking may be of interest: http://carbsanity.blogspot.com/2011/04/fatty-acid-trafficking.html
Post a Comment
Comment Moderation is ON ... I will NOT be routinely reviewing or publishing comments at this time..