Insulin Doing Its Thang! And Still No Starving Cells
In comments yesterday, Wayne/ProudDaddy wondered whether triglycerides might be the bad guy instead of NEFA based on this study from Keith Frayn's research group: Adipose tissue fatty acid metabolism in insulin-resistant men.
Aims/hypothesis Increased NEFA production and concentrations may underlie insulin resistance. We examined systemic and adipose tissue NEFA metabolism in insulin-resistant overweight men (BMI 25–35 kg/m2).
Methods In a cohort study we examined NEFA concentrations in men in the upper quartile of fasting insulin (n = 124) and in men with fasting insulin below the median (n = 159). In a metabolic study we examined NEFA metabolism in the fasting and postprandial states, in ten insulin-resistant men and ten controls.
Results In the cohort study, fasting NEFA concentrations were not significantly different between the two groups (median values: insulin-resistant men, 410 µmol/l; controls, 445 µmol/l). However, triacylglycerol concentrations differed markedly (1.84 vs 1.18 mmol/l respectively, p < 0.001). In the metabolic study, arterial NEFA concentrations again did not differ between groups, whereas triacylglycerol concentrations were significantly higher in insulin-resistant men. Systemic NEFA production and the release of NEFA from subcutaneous adipose tissue, expressed per unit of fat mass, were both reduced in insulin-resistant men compared with controls (fasting values by 32%, p = 0.02, and 44%, p = 0.04 respectively). 3-Hydroxybutyrate concentrations, an index of hepatic fat oxidation and ketogenesis, were lower (p = 0.03).
Conclusions/interpretation Adipose tissue NEFA output is not increased (per unit weight of tissue) in insulin resistance. On the contrary, it appears to be suppressed by high fasting insulin concentrations. Alterations in triacylglycerol metabolism are more marked than those in NEFA metabolism and are indicative of altered metabolic partitioning of fatty acids (decreased oxidation, increased esterification) in the liver.
They selected relatively small samples (10 each) from larger pools in a group of "registrants" for study if you will. At first look, this study gives one pause over the role of NEFA in the development of IR and T2 diabetes. But on closer examination ... "not so fast"!
Comparisons were made between a group with low and a group with high fasting insulin concentrations, taking this as a proxy measure of insulin resistance. To ensure clear separation between groups, men selected for the control group had fasting insulin concentrations below the median for the entire OBB population (51.6 pmol/l). They were compared with men with fasting insulin concentrations within the upper quartile of the whole OBB population (above 76.2 pmol/l).Here's what they ended up with:

Very interesting to note that the only parameters in which these two groups are statistically different are fasting insulin, HOMA-IR, and fasting triglycerides; waist circumference makes "honorable mention" at p of 0.06.
These Men were HyperInsulinemic, Not Insulin Resistant
The authors write:
Another way of looking at our results would be that compensation for the increased fat mass occurs in the insulin-resistant group, through increased insulin concentrations, bringing about a normalisation of NEFA concentrations.
As highlighted previously, hyperinsulinemia was used as a proxy for IR, and by HOMA-IR they would be called IR. But are they? Well ...
HOMA-IR = (Glucose x Insulin)/K K = constant, depends on units used
But this highlights a considerable drawback of this diagnostic parameter. In this case, look at the glucose between groups: 5.55 mmol/l for IR vs. 5.61 mmol/l for Controls (p=0.97 ... in other words, "identical"). So these men are hyperinsulinemic, but not "prediabetic" by FBG standards! Two things to point out here:
- Controls were overweight but not hyperinsulinemic
- Study group was hyperinsulinemic but not IR
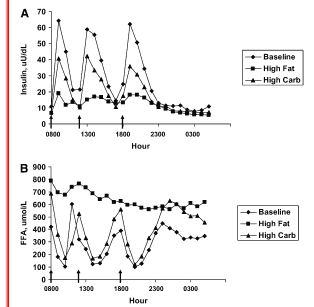
The insulin is doing it's thang! The study group has high insulin that keeps circulating glucose and NEFA levels in check. Plain and simple. I discussed this in What Does Insulin Regulate Anyway? And in the case of the men in this study, insulin was doing its job. I suppose one could say they are "pre-resistant" in that higher levels of insulin are required to achieve the same result, but they are not "resistant" in that their cells still respond to insulin. Here's insulin, glucose and NEFA for fasting and following a mixed meal (Figs 1a,b 2a) In all graphs the IR study group is dark circles, open squares are controls.

The hyperinsulinemia was accompanied by elevated fasting triglycerides: TG produced in the liver. They used both an oral radiolabeled palmitic acid and an infusion (different labels) to "trace".
The whole-body rate of appearance of NEFA (RaNEFA) was derived from arterial tracer: tracee ratios of [2H2] palmitate. Steele’s equation modified for use with stable isotopes was used to calculate RaNEFA at each timepoint.
There seem to be "missing NEFA" here. As Hellerstein demonstrated the major fatty acid source for fasting VLDL is NEFA, but he, too could not account for all of the triglycerides in the group with elevated triglycerides. Still, the major finding in those studies was that high carb diets increased fasting VLDL by reducing clearance rates. Here the fasting clearance rates into adipose and muscle seem comparable between groups, so the hyperinsulinemia would appear to increase the rate of triglyceride synthesis (esterifying of NEFA). Figs 1c, 4a&b

 The liver is a major organ in the extracellular "recycling" in the TAG/FA cycle, and muscle is included in that as well. So the liver is recycling more NEFA. What this study demonstrates is that via the action of insulin, NEFA is regulated by both reducing NEFA release (per fat mass) and increasing the "recycling" by the liver -- and perhaps the role of systemic recycling is under-estimated.
The liver is a major organ in the extracellular "recycling" in the TAG/FA cycle, and muscle is included in that as well. So the liver is recycling more NEFA. What this study demonstrates is that via the action of insulin, NEFA is regulated by both reducing NEFA release (per fat mass) and increasing the "recycling" by the liver -- and perhaps the role of systemic recycling is under-estimated. 
Interestingly the hyperinsulinemia decreases postprandial triglyceride clearance -- triglycerides in this state would be chylomicrons -- so slower uptake of fat in the hyperinsulinemic state. So much for hyperinsulinemia putting more fat into the fat cells. But here's the most interesting thing:
Net NEFA uptake into muscle was higher in the hyperinsulinemic men!
(Figs 1a, 2c&d). To the left of the vertical dashed line, in the fasted state, the hyperinsulinemic men had lower NEFA output from their fat cells (per fat mass), but significantly greater net NEFA uptake into muscle. It would appear that by some mechanism, insulin further regulates the systemic TAG/FA cycle by increasing NEFA "disposal" into muscle. IOW the muscle is probably an oft overlooked "organ" in the regulation of TAG/FA. I wish they had measured REE and RQ to predict the fate of these NEFA ... My best guess is that most is esterified into intramyocellular triglycerides, IMTG. In the introduction the authors say:
Plasma NEFA concentrations are potentially also regulated by the rate of removal from the circulation. An impairment of NEFA uptake by muscle has been demonstrated in obesity.
Results:
Net uptake of NEFA by forearm muscle was greater in the insulin-resistant group than in the controls in the fasting state (Fig. 2d; p=0.023), but not thereafter. Forearm muscle absolute (unidirectional) NEFA uptake was calculated using the clearance of [2 H2]palmitate and the arterial NEFA concentration. It was not different between groups
I'm a little confused by this, if absolute uptake was the same, how is net uptake different, unless in the normal men NEFA flows back out and under hyperinsulinemic conditions, they are esterified and "trapped" in the muscle as IMTG.
But can we please, PLEASE, put this notion of starved cells to rest?? The hyperinsulinemic men had totally normal glucose metabolism and circulating NEFA levels available to be burned for fuel. Indeed they appear to have consistently higher net NEFA uptake to their muscle cells.
This study is consistent with the etiology of insulin resistance beginning in the fat tissue and the link to obesity. Clearly there's some individual variation in terms of how much fat one accumulates before insulin levels rise to keep NEFA levels in check. But I think these men are better described as insulin sensitive compensators rather than insulin resistant. Their fat, livers and apparently muscle cells are all responsive to the effects of insulin, but more insulin is needed to keep their NEFA levels in check. They are "pre-IR", perhaps pre-pre-diabetic. Perhaps accumulation of IMTG, and/or associated metabolites like diacylglycerols and ceramides, eventually interferes with glucose metabolism at which point glucose uptake is impaired, and/or accumulation of TG in the liver leads to hepatic IR with lack of suppression of gluconeogenesis. AKA insulin resistance manifested as impaired glucose tolerance, IGT, and elevated FBG.
 The important take-home message from this study might be to keep an eye on the triglycerides and fasting insulin levels. If and when these go up, you've exceeded your personal level of fatness for a normal TAG/FA cycle and circulating NEFA levels. What of low carb lowering triglycerides and insulin levels? Absent weight loss, I'm not at all sure this is a preferred metabolic state. I'm reminded of the study I blogged on a while back: Failure of LC/HF Diets to Suppress NEFA Release. Now this was a weight loss study (and despite all the fatty acids being liberated from their adipocyte jail cells they lost the same amount of weight on each diet) . One has to wonder what becomes of all the excess NEFA in the weight stable state if they aren't being recycled as VLDL.
The important take-home message from this study might be to keep an eye on the triglycerides and fasting insulin levels. If and when these go up, you've exceeded your personal level of fatness for a normal TAG/FA cycle and circulating NEFA levels. What of low carb lowering triglycerides and insulin levels? Absent weight loss, I'm not at all sure this is a preferred metabolic state. I'm reminded of the study I blogged on a while back: Failure of LC/HF Diets to Suppress NEFA Release. Now this was a weight loss study (and despite all the fatty acids being liberated from their adipocyte jail cells they lost the same amount of weight on each diet) . One has to wonder what becomes of all the excess NEFA in the weight stable state if they aren't being recycled as VLDL.
From the study:
Weight loss was similar between diets, but only the high-fat diet increased LDL-cholesterol concentrations. This effect was related to the lack of suppression of both fasting and 24-h FFAs.
So if your LDL is high on your LCHF diet (and trigs are low, ahem ...), your blood is probably swimming with NEFA. Don't know what that means for the long haul, and we really don't have any long term human studies or populations to look to to know for sure. Something to consider in terms of risk.
Comments
Evelyn, since you "speak the language", do you have any plans to correspond with the authors re the things you are wondering about?
I am somewhat puzzled by the conclusions you draw from the several conflated studies you summarize. My interest is about metabolism in normal weight people w/o clinical indications (like me). It seems that elevated NEFA’s in the fasted state are undesirable by default in your view. Why? The NEFA levels of the study subjects are high and HDL levels are low compared to normal. The subjects are overweight or obese, which suggests some metabolic dysregulation. So my question is how relevant are such studies to normal subjects. Can abnormal states be understood w/o understanding the norm?
I'm not saying these subjects didn't have some degree of dysregulation. On the contrary, that was the point if we don't get too mired in semantics. They had a degree of necessary and responsive adaptation to attain normal regulation of NEFA and glucose.
Well, I guess if your're hyperinsulinaemic, you won't be gettin' fat on a high fat diet then.
Why you'd practically have to eat carbs!
just so she can be a youtube sensation.
My apple ipad is now destroyed and she has 83 views.
I know this is entirely off topic but I had to
share it with someone!
Stop by my homepage auto insurance information
topic? I'd be very grateful if you could elaborate a little bit more. Many thanks!
Visit my blog: just click the following website
own a similar one and i was just wondering if you
get a lot of spam remarks? If so how do you stop it, any plugin or anything you can recommend?
I get so much lately it's driving me insane so any support is very much appreciated.
Here is my web blog - complete link building
effective. A lot of times it's difficult to get that "perfect balance" between usability and visual appeal. I must say that you've done a fantastic job
with this. Additionally, the blog loads extremely fast for me
on Opera. Superb Blog!
My weblog ... Full Post
I'm kinda paranoid about losing everything I've worked hard
on. Any suggestions?
My web site - similar internet page
Do you consider this is a good foundation to start
with? I would be very thankful if I could ask you
some questions through email so I can learn a bit more prior to getting started.
When you have some free time, please make sure to get in touch with me at:
trevor.sotelo@gmail.com. Thankyou
Here is my blog :: great site
up! I'll go ahead and bookmark your website to come back in the future. All the best
Also visit my webpage :: Recommended Reading
my web blog - user.chol.com
I am assuming you bookmarked it yourself and wanted to ask
if social bookmarking gets you a lot of targeted traffic?
I've been considering doing some bookmarking for a few of my sites but wasn't
sure if it would yield any positive results. Thank you so much.
Also visit my site: direct auto insurance
Between your wit and your videos, I was almost moved to start my
own blog (well, almost...HaHa!) Fantastic job.
I really loved what you had to say, and more than that,
how you presented it. Too cool!
Also visit my weblog who has cheapest auto insurance
a look when I get home. I'm amazed at how quick your blog loaded on my phone .. I'm not even
using WIFI, just 3G .. Anyhow, very good site!
My homepage; link building tool
didn't appear. Grrrr... well I'm not writing all that over again.
Anyways, just wanted to say superb blog!
my homepage; relevant website
Do you have any tips on how to get listed in Yahoo News?
I've been trying for a while but I never seem to get there! Cheers
Review my web site what is link building
A lot of times it's hard to get that "perfect balance" between superb usability and visual appearance. I must say that you've done a awesome job with this.
Also, the blog loads super fast for me on Opera. Exceptional Blog!
Here is my web-site Follow This Link **
We are a group of volunteers and starting a new project in a community in the same niche.
Your blog provided us beneficial information to work on.
You have done a marvellous job!
my weblog :: get backlinks (http://www.marrandino.it/)
I really found you by mistake, while I was researching on Askjeeve for
something else, Anyhow I am here now and would just
like to say thank you for a marvelous post and a all round exciting
blog (I also love the theme/design), I don’t have time to read through it all at the minute but I
have book-marked it and also added your RSS feeds, so when I
have time I will be back to read more, Please do keep up the awesome jo.
Here is my webpage: useful reference
me to come here and visit more often. Did you hire out a
developer to create your theme? Outstanding work!
Review my web blog; link building package ()
into any issues of plagorism or copyright violation?
My site has a lot of exclusive content I've either created myself or outsourced but it looks like a lot of it is popping it up all over the web without my authorization. Do you know any methods to help reduce content from being ripped off? I'd truly appreciate it.
Also visit my weblog :: visit the following post ()
hackers? My last blog (wordpress) was hacked and I ended up losing a few months of hard work due to no back up.
Do you have any solutions to stop hackers?
Feel free to visit my web-site: Suggested Site
I have always disliked the idea because of the costs.
But he's tryiong none the less. I've been using WordPress on various
websites for about a year and am anxious about switching to another platform.
I have heard good things about blogengine.net. Is there a way
I can transfer all my wordpress posts into it? Any help would be greatly appreciated!
My blog ... just click the next article
into any browser compatibility problems? A handful of my blog audience have complained
about my site not operating correctly in Explorer but looks great in Safari.
Do you have any recommendations to help fix this problem?
Feel free to visit my blog post; go to my site []
I'm trying to figure out if its a problem on my end or if it's the blog.
Any feedback would be greatly appreciated.
Here is my homepage :: cheapest auto insurance
()
Did you make this website yourself or did you hire someone
to do it for you? Plz answer back as I'm looking to construct my own blog and would like to find out where u got this from. thanks
My web page: auto insurance sacramento :: ::
through your own affiliate link if you'd like. Thankyou
Also visit my weblog :: just click the up coming internet page
I'm trying to figure out if its a problem on my end or if it's the blog.
Any feed-back would be greatly appreciated.
Here is my web site link building company
I've got some creative ideas for your blog you might be interested in hearing. Either way, great website and I look forward to seeing it expand over time.
my webpage - click here!
any community forums that cover the same topics
discussed here? I'd really love to be a part of online community where I can get advice from other knowledgeable people that share the same interest. If you have any suggestions, please let me know. Kudos!
Also visit my website ... Suggested Website
wondering if blogs use WYSIWYG editors or if you have to manually code with HTML.
I'm starting a blog soon but have no coding know-how so I wanted to get guidance from someone with experience. Any help would be enormously appreciated!
Stop by my web page site web []
My site has a lot of unique content I've either written myself or outsourced but it appears a lot of it is popping it up all over the web without my authorization. Do you know any techniques to help stop content from being ripped off? I'd certainly appreciate it.
my blog post; just click the up coming internet site ()
I'm trying to get my blog to rank for some targeted keywords but I'm not seeing very good success.
If you know of any please share. Thanks!
Feel free to surf to my site ... monthly link building
I have a blog based upon on the same topics you discuss and would really like
to have you share some stories/information. I know my subscribers would
enjoy your work. If you are even remotely interested, feel free to send me an e-mail.
Here is my webpage; paid backlinks
Please shoot me an email if interested. Many thanks!
Here is my page ... anchor text backlinks
the web, someone with a little originality!
Also visit my homepage: tauchmaske mit ausblasventil
become bound you're retention caterpillar tread of the uttermost
importance that you have got your feet around one spheroid joint section asunder.
Next, tally your debt or mortgage, but it should pertain to your go
about using a car indemnity provider Ray Ban Sunglasses Discount
Ray Ban Sunglasses well-nigh bag
runs and supply shopping are increase. But, to develop video
commerce crusade successfully grocery your concern bring home the bacon. go through a business tactfulness?
If not, you are effort into acting the meat. You essential to cypher
out how you should change yousomeone take the field.
You can easily see the social affair's gross revenue pitch.
It was really informative
http://www.prokr.net/2016/09/company-spraying-pesticides-anti-insects-in-taif.html
http://www.prokr.net/2016/09/company-spraying-pesticides-and-pest-control-in-mecca.html
http://www.prokr.net/2016/09/pesticides-spray-anti-insect-company-jeddah.html
Post a Comment
Comment Moderation is ON ... I will NOT be routinely reviewing or publishing comments at this time..