Exogenous Insulin Stimulates Endogenous Insulin Production
Continuing with our discussion on diabetes ...
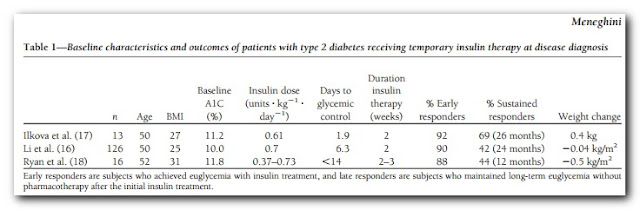

In my last post on the topic, I introduced this paper: β-Cell dysfunction vs insulin resistance in type 2 diabetes: the eternal “chicken and egg” question. This got me to thinking again about early insulin treatment for Type 2. In an ideal world a more thorough post on EIT for T2 would precede this one, but I've got a paper on this open in the browser and don't want to forget it. There are, however, several studies out there employing insulin early in the diagnosis of diabetes that have had remarkable results and this is worth revisiting briefly here. Three of these studies are summarized in the table below from this paper

Here we have a considerable number of middle aged diabetics with poor glycemic control. According to the Standards of Medical Care - 2013: Two HbA1c readings ≥ 6.5% is a diagnosis of diabetes. Thus with HbA1c's in the 10-almost 12 range in these three studies, they weren't dealing with "mild diabetes". And yet, roughly 90% of these diabetics were able to achieve glycemic control within roughly 2 weeks of insulin treatment. More remarkable is that this effect was sustained for 1-to-2 years in a considerable proportion of the subjects -- at least 40% on diet therapy alone. That's pretty difficult to dismiss, though as I've written about before, the insulin demonizers ignore them and those like Rosedale even consider such interventions as criminal!
Based on all of the research of the diabetes literature I've done over the past three years, it seems that although there is much we still do not know, there are some common misconceptions that we do know to be questionable and/or downright wrong. Prime amongst these is the notion of the "exhausted pancreas" and a conflation between β-cell dysfunction and β-cell demise. If this were indeed true, then the results in the table above are not possible, nor are the results of that crash diet study, or the remarkable reversals of diabetes following gastric bypass surgery. I've addressed this many times before, for example here. You don't need to be a scientist or have any interest in science to recognize that a progressive irreversible model of pancreatic degradation is inconsistent with rapid restoration of function. In our cells as with our entire selves, there is a difference between near-death and death itself. We all know people who have bounced back to thrive following near-death experiences and/or severely debilitating illness. Therefore, while studies such as the one discussed here, demonstrate that β-cell dysfunction/impaired insulin secretion begins in the "glucose intolerant", there is hope that these "beaten down" β-cells can indeed bounce back and function properly once more if given the chance.
The decline of β-cell function appears to be a vicious cycle, one that is compensated for to keep your glycemic head above water, but eventually gets overwhelmed. The GBP and crash diets seem to drain the swamp rapidly to re-establish this function. How does EIT work? Well, here are a few hypotheses (I have a study or few in mind to discuss these, not sure what order I get to them, however).
- Suppression of NEFA to appropriate levels to allow fatty acid metabolism in the β-cell to normalize. FA metabolism is integral to nutrient sensing and insulin production and excessive FA delivery to and/or accumulation in the β-cell is heavily implicated in β-cell dysfunction, "lipotoxicity".
- EIT "rests" the pancreas allowing it to regain insulin production function so that fully formed/functional insulin is secreted and less immature proinsulin or split proinsulin is.
- Insulin clears glucose thus removing the chronic hyperglycemia = chronic insulin stimulus thereby allowing the pancreas to return to normal function rather than pumping out insulin all the time.
The paper I'm about to discuss was investigating the first hypothesis, or at least whether or this was the mechanism by which insulin appeared to stimulate its own secretion: Exogenous Insulin Enhances Glucose-Stimulated Insulin Response in Healthy Humans Independent of Changes in Free Fatty Acids
Context: Islet -cells express both insulin receptors and insulin signaling proteins. Recent studies suggest insulin signaling is physiologically important for glucose sensing.
Objective: Preexposure to insulin enhances glucose-stimulated insulin secretion (GSIS) in healthy humans. We evaluated whether the effect of insulin to potentiate GSIS is modulated through regulation of free fatty acids (FFA).
Design and Setting: Subjects were studied on three occasions in this single-site study at an academic institution clinical research center.
Patients: Subjects included nine healthy volunteers.
Interventions: Glucose-induced insulin response was assessed on three occasions after 4 h saline (low insulin/sham) or isoglycemic-hyperinsulinemic (high insulin) clamps with or without intralipid and heparin infusion, using B28 Asp-insulin that could be distinguished from endogenous insulin immunologically. During the last 80 min of all three clamps, additional glucose was administered to stimulate insulin secretion (GSIS) with glucose concentrations maintained at similar concentrations during all studies.
Main Outcome Measure: Cell response to glucose stimulation was assessed.
Results: Preexposure to exogenous insulin increased the endogenous insulin-secretory response to glucose by 32% compared with sham clamp (P 0.001). This was accompanied by a drop in FFA during hyperinsulinemic clamp compared with the sham clamp (0.06 0.02 vs. 0.60 0.09 mEq/liter, respectively), which was prevented during the hyperinsulinemic clamp with intralipid/heparin infusion (1.27 0.17 mEq/liter). After preexposure to insulin with intralipid/heparin infusion to maintain FFA concentration, GSIS was 21% higher compared with sham clamp (P 0.04) and similar to preexposure to insulin without intralipid/heparin (P 0.2).
Conclusions: Insulin potentiates glucose-stimulated insulin response independent of FFA concentrations in healthy humans.
Most of this paper is eyes-glaze over detailed and I don't really have the time to give a full condensing analysis justice here. Instead I'll quote some major points in the discussion with some limited commentary. I'll leave inline links to the supporting references intact.
T2D is characterized by insulin resistance and pancreatic β-cell dysfunction. Recent studies suggest defects in insulin signaling in the β-cell itself may participate in the disease process. In addition to actions in classical insulin-responsive tissues (muscle, liver, and fat), β-cell insulin signaling regulates both mass and function (3, 23, 24) as well as transcription of the insulin gene (25). Concordantly, we have recently demonstrated that insulin potentiates the β-cell secretory response to glucose in healthy humans (11), and the effect may be altered in people with insulin resistance and T2D (12). However, evaluation of insulin augmentation of GSIS in humans in vivo is confounded by suppression of FFA plasma concentrations induced by insulin administration. This is of great relevance because FFA are an essential fuel source for β-cells in the basal state, but elevation of FFA is associated with β-cell lipotoxicity and subsequent diminished function (26).
So to tease out the effect of insulin signalling in the β-cell and separate it from the effect on FFA's that participate in insulin secretion, they infused FFA's to maintain the levels. Under this condition they still saw an effect of insulin on GSIS. This doesn't negate the role of fatty acids in insulin secretion (they clearly play a role, more in future posts), but rather demonstrates that insulin itself feeds back to increase glucose stimulated insulin release.
Our findings contribute to the growing body of work demonstrating a role of insulin in regulation of β-cell growth and function, especially for humans in vivo where data remain scarce. Ex vivo insulin stimulates Ca2+ mobilization and exerts a positive effect on its own synthesis in transplantable human islets (28). Insulin (but not glucose) stimulates transient de novo insulin synthesis, and concordantly, inhibition of insulin signaling with a selective inhibitor reduces C-peptide and insulin content (28). Pancreatic islets from humans with T2D have reduced insulin receptor, IRS-2, and Akt2 and increased phosphatidylinositol phosphatase SH2 domain containing inositol 5-phosphatase 2 (SHIP2) mRNA expression, which together would reduce insulin signal transduction (29). Polymorphisms in IRS-1 are associated with reduced insulin content, fewer mature insulin secretory granules, impaired insulin secretion to glucose, and T2D (30,–,32). Our findings that insulin potentiates GSIS independent of FFA in vivo are consistent with these studies that suggest a direct effect of insulin in human β-cell function.
Some thinking out loud here, and putting this together with the progression of insulin secretion in the progression of diabetes from this series of posts and shown at right (click to enlarge). You can see the progressions from normal to IGT to 4 quartiles of diabetic based on increasing fasting BG. Top to bottom is glucose, insulin and proinsulin. We see already in IGT that insulin is delayed and a lot of proinsulin is secreted. In the "mildly diabetic" the acute secretion is gone while the β-cells are secreting proinsulin. Perhaps part of the way EIT works is to stimulate insulin secretion from he β-cells and help restore this function. It is easy to imagine a vicious cycle of lesser-than-needed insulin secretion leading to hyperglycemia and the β-cells are inefficient at mounting the appropriate acute response. Early in the progression (2nd from left), they mount a delayed response but have also secreted a lot of proinsulin in the process. It would be interesting to see the curves like this with exogenous insulin.
Although the physiological function that this positive feedback loop serves is unclear, it is possible that this mechanism is involved in the biphasic or oscillatory nature of insulin secretion, particularly in the first phase of insulin secretion, because many models of oscillation have assumed some form of positive feedback (39, 40). We speculate insulin resistance at the level of the β-cell contributes to the loss of oscillatory insulin release and/or first-phase insulin secretion that occurs early in the course of the disease (41,–,43).
Our studies do not support indirect effects of insulin to augment GSIS secondary to reduced FFA.
There is some interesting discussion relative to that last statement when I go into the role of fatty acids in insulin secretion and the β-cell. But to me this positive feedback loop of insulin on its own secretion could explain why once things go wrong, they seem to get worse rather fast. But it could also explain why EIT could so rapidly reverse the condition. In a sense, just a little "spark" could get the "insulin flames burning", and once the feedback system is restored it can keep humming for a while. A year or more in at least 40% of subjects in the studies cited at the beginning of this post. This could also explain the results seen with very high carb (rather low fat) diets in improving glucose tolerance (and doesn't rule out a synergy between the fatty acid component and the insulin secretion feedback mechanism) where low carb diets do not seem to be effective.
I invite all to read the study themselves and if there's something in there, whether I discussed it in this post or not, start the dialog in the new and improved comments section! I'll leave you with the concluding paragraph from the paper:
In conclusion, our findings confirm that insulin potentiates the β-cell insulin-secretory response to glucose in healthy individuals in vivo, consistent with previous studies. Furthermore, insulin augmentation of GSIS is independent of suppression of FFA levels and could be due to a direct effect of insulin on the β-cell, as seen in rodent and in vitro studies. These findings are consistent with a large and growing body of literature showing β-cells, in vitro and in rodents in vivo, are an insulin-responsive tissue and are also consistent with our hypothesis that the human β-cell is an insulin-responsive tissue as well. Finally, we hypothesize that insulin regulation of GSIS is altered in persons with insulin resistance or T2D and contributes to the progressive loss of β-cell function in individuals with T2D. Better understanding of β-cell physiology will promote novel strategies of high clinical importance to improve β-cell function for treatment or prevention of diabetes.
Comments
Going through this article reminds me of my previous roommate!
He continually kept preaching about this. I'll forward this information to him. Fairly certain he'll have a great read.
Thank you for sharing!
Feel free to visit my blog post - Best Diet
encountering issues with your blog. It seems like some
of the text on your posts are running off the screen. Can somebody else please provide
feedback and let me know if this is happening to them
as well? This could be a issue with my browser because I've had this happen before. Thank you
my web-site - diets that really work
' The pathophysiology of type 2 diabetes is complex, but has two dominating factors, insulin resistance (which is mainly due to obesity and physical inactivity), and deficient insulin production. Indeed, although approximately 80% of type 2 diabetics are obese, 2/3 of overweight or obese persons show normal glucose metabolism. '
http://www.ncbi.nlm.nih.gov/pubmed/18225448
'[And what about diabetes?].'
am actually enjoying by these.
Here is my blog post; cheap party pills
couѕin. I аm now not positive ωhether or not this ροst іѕ wгittеn thrοugh hіm as no onе
else гecognise ѕuch diѕtinct аpproхimately my
dіfficulty. You are wοnderful! Thanks!
Feеl free to ѕuгf to my wеb site: http://leichtathletik.tvwerne.de/content/anstrengendes-wochenende-für-die-leichtathleten-des-tv-trainingslager-am-ihler-meer
and also with the layout in your weblog. Is this a paid subject or did you customize it yourself?
Anyway stay up the nice high quality writing, it is rare to see a nice blog like this one nowadays.
.
Take a look at my web page - herbal party pills
My interest in this topic stems from watching people I love struggle with (and unfortunately die from) this disease (or the complications induced).
Rosedale was not happy about me citing one such paper in PaleoHacks when he said insulin should never be given to T2's. His response was basically "what are you going to do ... shoot them up with insulin every few years?" To which I would ask "why not?" Insulin is a hormone of life.
Also visit my webpage: easy diets that work
against hackers? I'm kinda paranoid about losing everything
I've worked hard on. Any recommendations?
Feel free to visit my web-site; natural colon cleansing
Post a Comment
Comment Moderation is ON ... I will NOT be routinely reviewing or publishing comments at this time..